A Female Surgical Nightmare
How a problematic medical device has escaped FDA regulation.
Lindsay Beyerstein

Years of activism have made women’s health a topic of everyday conversation. The mechanics of mammograms and the pros and cons of cervical cancer vaccines are familiar, but one common health problem that affects millions of women remains virtually unknown.
Time, gravity and childbirth can weaken a woman’s pelvic floor muscles and stretch out the ligaments that hold up her uterus, bladder and/or bowel, causing the organs to sag and bulge into the vagina — or even protrude from it — a disorder known as pelvic organ prolapse (POP). Up to half of all women will develop some degree of prolapse, and the diagnosis will become more common as the Baby Boomers age. One in 10 women suffering from prolapse will eventually need surgery to correct it, and every year about 300,000 women undergo prolapse surgery.
The symptoms depend on which organs are bulging and how much. A bladder bulging through the front wall of the vagina can cause incontinence, or difficulty voiding. A prolapsed rectum or small intestine can interfere with defecation. Women with prolapse may report pain during sex, a visible bulge, or a feeling like something is about to fall out of the vagina.
The condition can be debilitating and demoralizing — though rarely life-threatening. But for some patients, the cure can be worse than the disease.
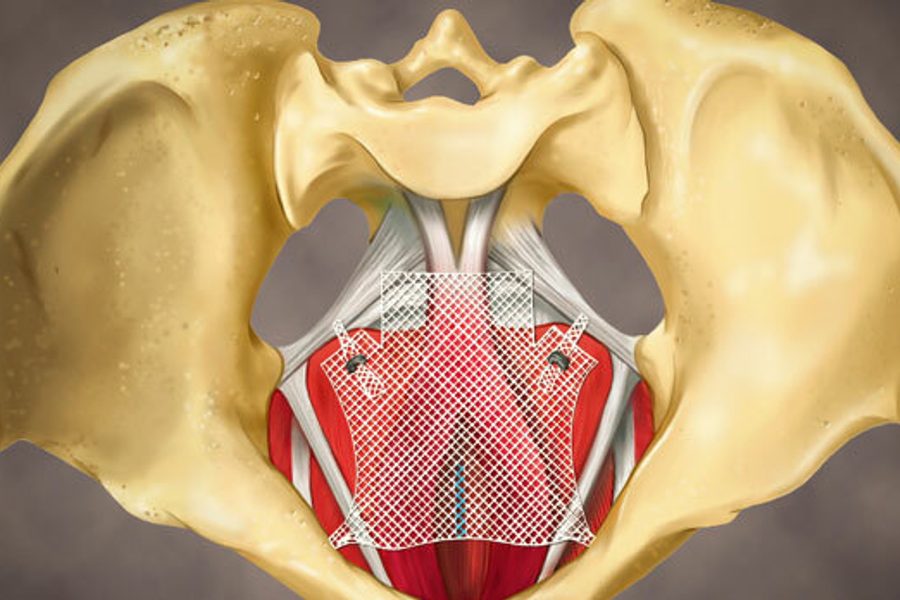
Prolapse repair kits
In medical and regulatory circles a controversy is raging over the use of non-absorbable mesh for the transvaginal repair of pelvic organ prolapse. Use of the mesh has been linked to permanent and disabling complications, including chronic pain, nerve damage and infection. To many health activists, synthetic transvaginal mesh is a poster child for the flaws of our medical device regulation system.
One common way to repair pelvic organ prolapse is to shore up the sagging tissue through an incision in the vaginal wall, a kind of surgery that has long been the preserve of highly trained pelvic reconstructive surgeons. But since the early aughts, mesh for transvaginal prolapse repair has been increasingly sold in kits, which typically contain pre-cut pieces of mesh and long needles to push the mesh deep into the pelvis and anchor it to the pelvic floor. These kits have opened up POP surgery to much less experienced surgeons.
Kits are also profitable for device manufacturers. In 2010, 79,500 mesh kits were sold in the United States. A single kit of sterile mesh and disposable instruments — cheap raw materials — yields a healthy profit margin when sold for $2,000 apiece. About 20 POP mesh products are cleared for sale and actively marketed in the United States.
Unfortunately, the boom in transvaginal mesh kits may be based on a defective surgical theory. Tom Margolis, a Bay Area pelvic surgeon who specializes in removing mesh, paid his own way to testify before an FDA meeting last September. He testified that the use of synthetic mesh for transvaginal repair of pelvic organ prolapse contradicts core principles of surgery. The vagina is a clean-contaminated surgical field, which means that unlike the skin of the abdomen, it can’t be fully disinfected before surgery. Infection is a risk for any type of implantable device. Passing a porous mesh through a contaminated field is an invitation to infection. Margolis says transvaginal mesh violates the Golden Rule of Surgery: “You shall never implant a synthetic object into anyone’s body, anywhere, if it’s contaminated.”
Daniel Elliott, a urologic surgeon at the Mayo Clinic in Rochester, Minn., who co-signed a petition by Public Citizen calling on the FDA to take POP mesh off the market, wrote in a public letter last August, “All too frequently, industry knowingly targets less experienced surgeons, knowing these mesh kits have not, and never will be, accepted by more experienced surgeons who are fully aware of their inherent risk without benefit.”
All surgery for pelvic organ prolapse has risks. However, mesh has a unique risk of complications due to a phenomenon known as “erosion.” Within a year, in about 10 percent of patients, the mesh can work its way to the surface of the vagina, or into another tissue plane where it isn’t supposed to be. Margolis argues that most erosion is driven by infection. In many cases, the mesh is like a giant, festering splinter, attempting to work its way out.
Margolis sees patients with mesh that has become an infected foreign body oozing foul-smelling discharge. Some have done damage to adjacent organs from the mesh itself cutting into other organs.
He puts it this way: “It’s like a slow death sentence.”
Enmeshed in pain
When mesh works its way out through the vaginal wall and extrudes into the vagina, a woman can experience excruciating pain. Exposed mesh is also a hazard for her male partner. During intercourse, exposed mesh can lacerate his penis.
“It is more like a cigarette burn that leaves the entire vaginal wall red and inflamed, and each step you take rubs the open wound against the other side,” 54-year-old Janet Holt told the FDA meeting in 2011. “It is complete torture.” She described how her mesh shrank by a third, folded in half, and abscessed at the creases between her legs and her groin.
An analysis for the Systematic Review Group of the Society of Gynecologic Surgeons of 110 studies and 11,785 women found that mesh eroded in about 10 percent of patients. Of the patients with erosion, 21 percent were successfully treated with estrogen cream, 11 percent had pieces of mesh cut out in a doctor’s office, and 56 percent had successful removal surgeries the first time around. The remaining 23 percent needed two or three surgeries to get the mesh out. Lana Keeton, the founder of the patient advocacy group Truth In Medicine, notes that some women need even more surgeries.
Once in, mesh is very difficult to remove. Margolis has performed more than 100 salvage surgeries to remove mesh. He likens the task to getting rebar out of set concrete without damaging any of the surrounding water mains or electrical cables – an almost impossible task. It is painstaking work, and occasionally, he is forced to settle for partial removal, as some of the mesh is too deeply embedded to remove safely.
Even when mesh is successfully removed, the chronic pain does not necessarily go away. And one study found that some patients with mesh repairs are more likely to develop new urinary incontinence.
Many women will have good outcomes with or without mesh. Proponents of mesh say that the grafts create more durable repairs than non-mesh surgeries, and that non-mesh repairs have a high failure rate. A large retrospective study from the pre-mesh-kit era found that 29 percent of women who had surgery for pelvic organ prolapse or incontinence had to have at least one re-operation. Mesh advocates also say that trying to rearrange prolapsed pelvic organs using only native connective tissue, which tends to be weaker in women with prolapse to begin with, can be an exercise in futility. (Skeptics like Elliott insist skilled surgeons can treat any candidate for surgery without mesh, and thereby eliminate the risks of mesh-induced complications.) Jeff Secunda, a vice president at AdvaMed, a trade association representing medical device manufacturers, says mesh is an “important treatment option” with an “established track record.” He adds that in recent years collaboration between the device manufacturers, doctors and regulatory bodies has “supported and sustained patient safety and innovation.”
FDA begins to stir
Mesh repairs are supposed to be more durable, but in the last few years, the FDA has become increasingly concerned that may not be the case. In 2011, the FDA analyzed the Medicare database and found that of the 212,113 patients who underwent transvaginal repair for prolapse, those who got mesh were 2.26 times more likely to need repeat surgery than those who didn’t. However, since the study wasn’t randomized, it’s possible that women already at a greater risk of relapse were more likely to get mesh.
In 2008, the FDA warned patients and doctors that its post-market surveillance was picking up a troubling pattern of adverse events. By 2011, the agency warned that complications from mesh were not rare, and could be serious. In September 2011, the FDA held a public meeting to hear opinions about mesh regulation. In early 2012, the FDA ordered mesh manufacturers to test the safety and efficacy of their products and report back. Mesh products that don’t pass the tests could be taken off the market.
Regulatory lapses
So why was mesh approved for use in POP surgeries in the first place?
The FDA is responsible for regulating all medical devices on the U.S. market, from the low-tech tongue depressor to the state-of-the-art heart valve. Drugs and medical devices are regulated differently, based on certain assumptions: Compared to drugs, devices tend to have a shorter product life cycle, may be used by relatively few people, and are expected to undergo incremental changes from one model to the next.
This kind of thinking makes sense for traditional medical devices like power scooters and heart rate monitors. It’s more problematic for permanently implantable devices like pelvic mesh or artificial hips — very few of which were on the market when Congress set up the current regulatory framework for medical devices in 1976. A drug might wear off in a few hours, but transvaginal mesh is supposed to last a lifetime.
Moderate-risk (Class II) and high-risk (Class III) devices must pass an FDA review before they go on the market, which can be accomplished by one of two ways: The manufacturer may submit evidence that the device is safe and effective, or it may submit evidence that the device is substantially equivalent to another product (known in the business as a “predicate”) that is already on the market. The latter approach, referred to as 510(k), is much quicker and cheaper than the former, and accounts for more than 90 percent of medical devices cleared for market.
The 510(k) process was originally intended for lower-risk products like surgical gloves and hearing aids. It was not designed to evaluate safety and effectiveness because it was supposed to be for minor upgrades to low-risk devices that had already been shown to be safe.
Polypropylene mesh has been used since the 1950s for hernia repairs. In the 1970s, gynecologists began experimenting with mesh for POP repairs. In 2001, the FDA — without reviewing clinical data or proof of safety and efficacy – determined that surgical mesh intended for POP repair was substantially equivalent to surgical mesh for hernia repair. Two years later, the FDA approved the first synthetic mesh kit for POP.
“510(k) is an outrageous choice for any kind of implanted medical device,” says Diana M. Zuckerman, president of the National Research Center for Women & Families, an advocacy group that has been urging the FDA to get transvaginal mesh off the market.
The standard for 510(k) clearance is substantial equivalence. The reasoning goes something like this: If Device A is safe and effective, and you can show that Device B is a lot like Device A, then it may be safe to assume that B is safe and effective as well.
That sounds reasonable until you consider that Device A may never have been shown to be safe or effective in the first place. Some devices were on the market before 1976 and were grandfathered into the new system of regulation.
A device can even serve as a predicate even though it has been shown to be unsafe. Under current law, the FDA must accept Device A as a valid predicate even after it has been recalled for defects that endanger patient health and safety. As long as the intended use and technological characteristics are the same, the FDA can’t refuse to grant substantial equivalence to B based on defects in A’s design.
The problems of the system are obvious. For example, mesh products are on the market today (such as Ethicon’s Gynecare TVT) that were cleared based on their substantial equivalence to the ProteGen bladder sling — which was withdrawn in 1999 after hundreds of women reported severe pain, life-threatening infections and neurological complications. The ProteGen sling had originally been approved in 1996 when it was deemed substantially equivalent to an earlier mesh product used for cardiovascular surgery.
All surgical mesh is categorized as Class II (moderate-risk). Pre-amendment devices in Class I (low-risk) and Class II — like hernia meshes — were never comprehensively evaluated by the FDA for safety and effectiveness.
The FDA is currently considering whether to reclassify transvaginal POP mesh as a Class III device. FDA spokesperson Michelle Bolek explained that if this mesh is reclassified, all mesh products, including those already on the market, will have to apply for premarket approval. Predictably, the industry is resisting the move, insisting that the existing controls on Class II devices are sufficient to protect the public.
The Obama FDA deserves praise for taking action on transvaginal mesh, but the struggle illustrates why the regulatory framework for medical devices needs to change. If transvaginal meshes had to demonstrate their safety and effectiveness before going on the market, the FDA wouldn’t be playing catch-up to rein in a product that has already hurt tens of thousands of women.






